MPeaks
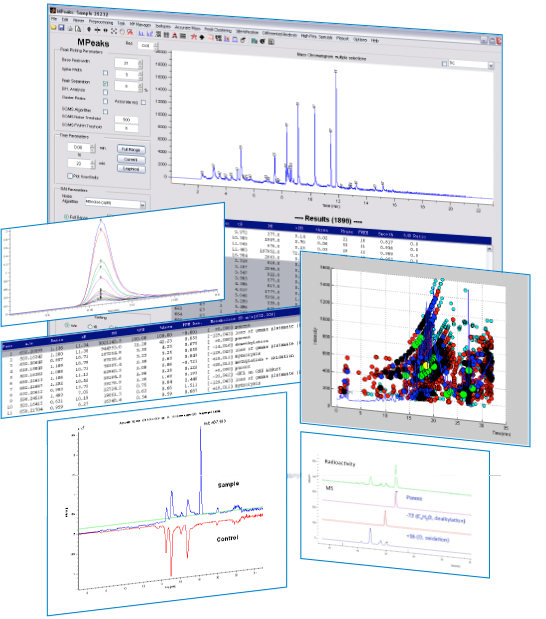
The core of the MPeaks Module are a number of high performance Peak Picking algorithms for LC/MS and GC/MS. MPeak’s Peak Picking is extremely fast, easy to use and can be applied at any resolution and sensitivity. Peak Picking focuses on detection of real significant chromatographic peaks from all Extracted Ion Currents (EIC).
MPeak’s results are clearly presented in complete peak tables, including many diagnostics on each detected peak. Results from MPeaks are the basis for many subsequent analysis tools found in the MPeaks Module, e.g. Differential Analysis and Identification.
Mpeaks Features:
Fast and sensitive Peak Picking/Deconvolution for LC/MS and GC/MS data sets.
- Determine peak widths, peak area/height, charge states and optionally convert all peaks to mono-isotopic peaks.
- Clustering of peaks: combine GC/MS fragment ions into single clusters of co-eluting peaks. For LC/MS, group multiple charged ions, fragments and adducts into one group.
- Optimize Peak Picking settings using one single file. Next, run MPeaks on a large number of samples and output the results to a combined peak picking list to be used for Comparative and Statistical Analysis.
Comparative / Differential Analysis
- Quickly find all differences between sample and control using Differential Analysis. Differential Analysis uses a shift tolerant algorithm to detect significant differences at any resolution.
- Check which peaks from Differential Analysis were not detected and measured by MSMS. Using the differential list as an inclusion list re-analyze the sample, but now only for the peaks of interest.
Automatically Identify all your detected peaks:
- Metabonomics: link to existing or user created Metabolite ID data bases.
- Molecular Formula Finder: finds all formulas for all peaks in the table based on accurate mass. Includes Isotope Matching and viewing of simulated MS spectrum for each formula.
- Proteomics: link to Mascot results files. Determine which interesting peaks were missed by MSMS by comparison of Full Scan peak lists and MSMS results.
- Drug Metabolite Profiling: use pre-defined Biotransformation rules to identify potential metabolites from all detected peaks. Can be used in combination with Mass Defect Filtering.
- GC/MS: perform identification of raw or deconvoluted MS spectra of detected peaks by linking with NIST MS libraries.
Apply automatic full data set pre-processing and filtering to enhance Peak Picking:
- Smoothing
- Mass Defect Filter (MDF) to remove non-relevant background ions.