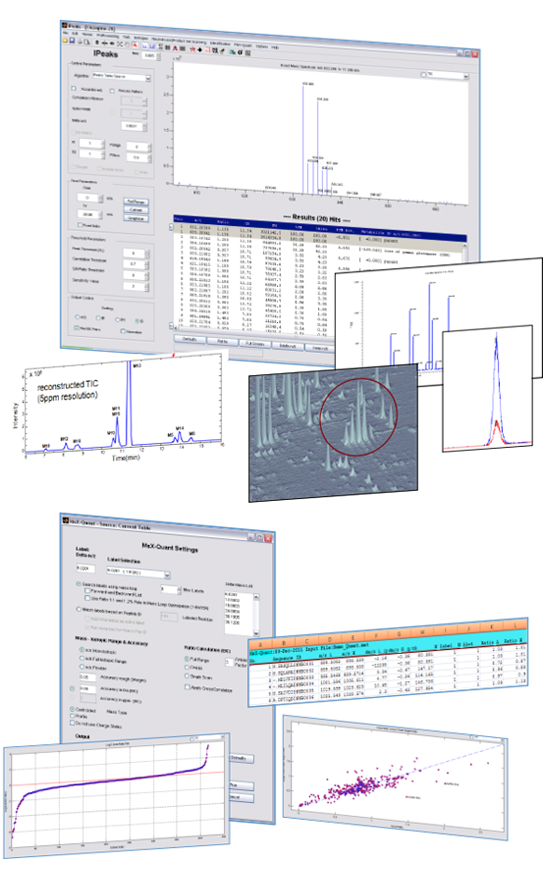
IPeaks
The IPeaks module of the MsXelerator software contains advanced algorithms to detect and quantify peaks based on isotopic patterns. The origin of these peaks can be natural, e.g. in case of compounds containing Chlorine or Bromine like in Drug Metabolite Profiling, or they can be based on mixing of un-labeled and labeled samples in specific ratios (Quantitative Proteomics, SILAC).
Peak Detection based on Isotopic Patterns:
Given the accurate mass difference and expected ratio between light and heavy isotopes, two different algorithms can be used for isotopic peak detection: IPF I and IPF II. IPF I is mainly used for lower resolution instruments and is based on matching Extracted Ion Currents (EIC) while IPF II runs directly on the raw high resolution mass spectra.
Results of IPF I and IPF II are clearly presented in summary tables listing all features of the detected peak pairs and can be easily viewed and compared. Ratios are calculated based on accurate peak integration of the full 2D peak distribution or based on accurate peak heights.
Peak Quantitation in Proteomics:
If you need relative quantitation of all your peaks and precursor ions are already known, use MsX-Quant, included in IPeaks as a special algorithm.
Ipeaks Features:
Drug Metabolite Profiling:
- IPeaks contains pre-defined Isotopic Patterns for: Cl, Cl2, 13C, 14C, Br, GSH, KCN. Create and store user defined isotopic patterns.
- Reactive Metabolite Profiling: detect reactive metabolites based on GSH or KCN trapping assays. IPF II will detect all your peaks at the lowest levels in less than a minute.
- Automatically correct results by comparisons against control sample.
- Use Mass Defect Filtering as a preprocessing option.
- Automatically link results from IPF II with our Neutral Loss scanning algorithm (IPF III), e.g. in case of GSH assay, all MSMS scans are checked for the loss of 129 Da.
- Metabolite Identification: identify detected peaks based on common Biotransformation rules.
- Molecular Formula Finder: finds all formulas for all peaks in the table based on accurate mass. Includes Isotope Matching and viewing of simulated MS spectrum for each formula.
- High Resolution Post Processing: make use of our High Resolution Post Processing tools to remove possible false positives: remove very narrow peaks, only keep peaks also having a 13C pattern, remove higher charged peaks, compare against one or more control samples, remove adducts, keep only mono-isotopic peaks, search for combinations of isotopic patterns, e.g. GSH in addition to a Cl pattern.
Quantitative Proteomics: MsX-Quant:
- Use MsX-Quant to quantify SILAC based experiments based on Full Scan Data using three different procedures for generating a basic precursor list:1. Import Mascot result files. Extract precursor and other basic information to build mass list for quantitation. Optionally filter Mascot results before starting quantitation. 2. Import User generated Excel or Text file containing m/z values and sequence information for all peptides of interest. 3. Let MPeaks do the Peak Picking and perform quantitation with no sequence information. The best matching heavy isotope is found by an iterative procedure.
- Pre-defined Isotopic Patterns: SILAC, ICAT, ECAT, MIDAR, 16O/18O.
- Easy viewing, plotting and filtering of your results. Includes error analysis to obtain reliable results.
- Create user defined isotope patterns to detect peaks of interest for special experiments, e.g. binominal patterns.
- Use our fast 14N/15N algorithm without knowing the peptide sequence. The algorithm detects co-eluting pairs using an iterative procedure